Sickle cell disorders: Understanding the Genetics, Symptoms, Complications, and Treatment
Introduction
Sickle cell disorders (SCD), also known as sickle cell anemia, is a genetic blood disorder that affects millions of people around the world. It is a lifelong condition characterized by the presence of abnormal hemoglobin, a protein in red blood cells that carries oxygen. In this article, we will delve into the genetics, symptoms, complications, and treatment of sickle cell disease.
Genetics of Sickle Cell Disease:
Sickle cell disorders is inherited in an autosomal recessive manner, meaning that an individual must inherit two abnormal hemoglobin genes, one from each parent, to develop the disease. The abnormal hemoglobin gene responsible for SCD is known as HbS.
- If an individual inherits one HbS gene and one normal hemoglobin gene (HbA), they have a condition called sickle cell trait (SCT). SCT carriers are usually asymptomatic and can pass the HbS gene to their offspring.
- If both parents carry the HbS gene, there is a 25% chance with each pregnancy that their child will inherit two HbS genes, leading to sickle cell disease.
Symptoms of Sickle Cell Disease:
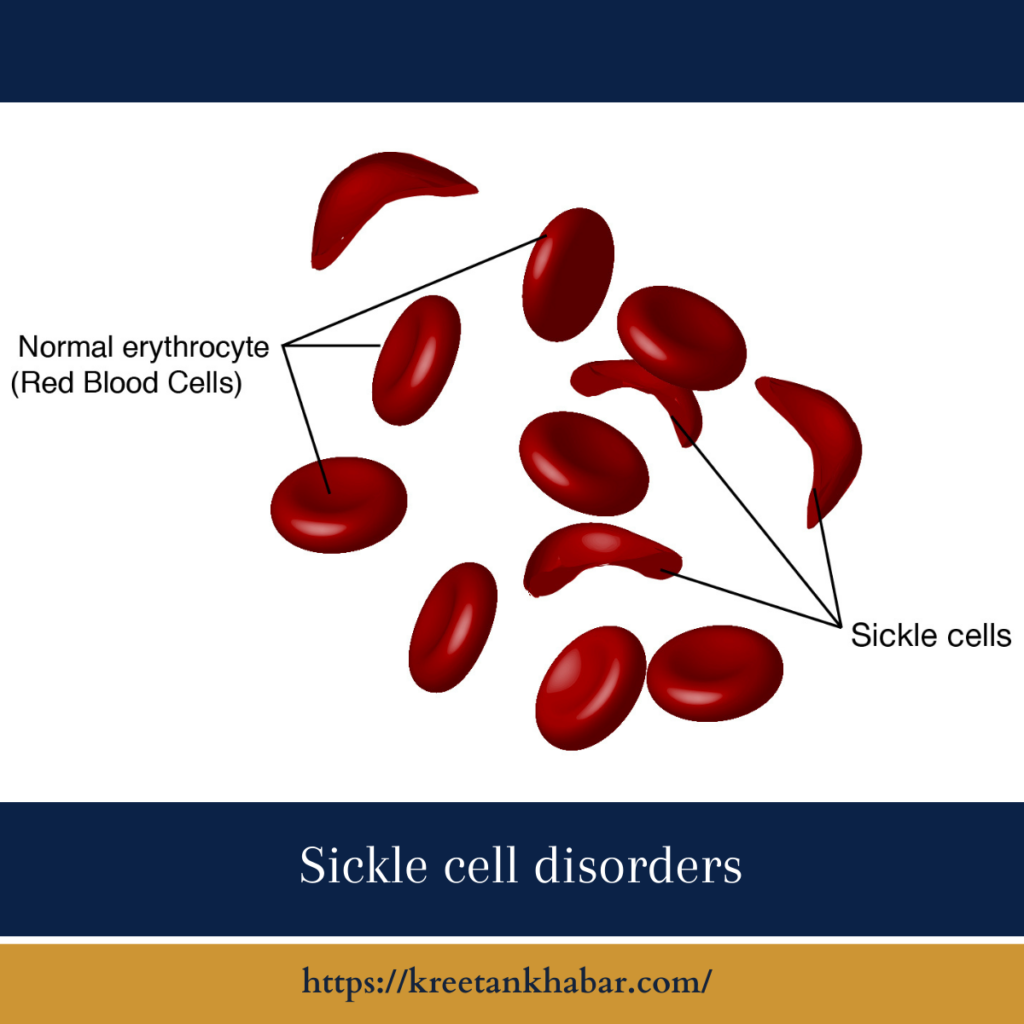
The hallmark symptom of Sickle cell disorders is the presence of sickle-shaped red blood cells, which are less flexible and can easily block blood vessels, leading to a wide range of symptoms. Common symptoms and complications include:
- Pain Crises: SCD patients often experience episodes of severe, excruciating pain, known as pain crises. These crises can affect various parts of the body and may require hospitalization and strong pain medications.
- Anemia: Sickle cells are fragile and have a shorter lifespan than normal red blood cells, leading to anemia. Anemia can cause fatigue, weakness, and pallor.
- Jaundice: The breakdown of sickle cells can release bilirubin, causing jaundice (yellowing of the skin and eyes).
- Hand-Foot Syndrome: Painful swelling of the hands and feet, especially in young children with SCD.
- Infections: SCD patients are more susceptible to infections, particularly those caused by encapsulated bacteria like Streptococcus pneumoniae.
- Organ Damage: Repeated episodes of sickle cell-related blockages can lead to organ damage, including damage to the spleen, kidneys, lungs, and bones.
- Stroke: SCD patients, particularly children, have an increased risk of stroke due to the impaired flow of blood to the brain.
- Priapism: Painful, prolonged erections can occur in males with SCD.
here are key points about the symptoms of Sickle cell disorders(SCD):
Symptoms of Sickle Cell Disease:
- Pain Crises: SCD is often characterized by recurrent and severe pain episodes, known as pain crises or vaso-occlusive crises. These crises can occur suddenly and affect various parts of the body, including the chest, abdomen, limbs, and joints. The pain is typically intense and can last for hours to days.
- Anemia: SCD leads to chronic hemolytic anemia, where red blood cells are destroyed faster than they can be produced. This anemia can cause fatigue, weakness, and pallor (pale skin).
- Jaundice: Hemolysis (destruction of red blood cells) releases bilirubin, a yellow pigment, into the bloodstream, leading to jaundice. Jaundice is characterized by yellowing of the skin and the whites of the eyes.
- Hand-Foot Syndrome: This condition, primarily seen in infants and young children with SCD, involves painful swelling of the hands and feet. It can be one of the early signs of the disease.
- Frequent Infections: SCD impairs the immune system’s ability to fight infections. Individuals with SCD are more susceptible to bacterial infections, particularly those caused by encapsulated bacteria like Streptococcus pneumoniae.
- Delayed Growth and Development: Children with SCD may experience delayed growth and development due to chronic anemia and illness.
- Enlarged Spleen (Splenomegaly): The spleen can become enlarged and overactive in individuals with SCD, leading to an increased risk of infection and the removal of damaged red blood cells.
- Organ Damage: Repeated episodes of blocked blood vessels (vaso-occlusion) can lead to organ damage over time. This can affect various organs, including the spleen, liver, kidneys, lungs, and bones.
- Stroke: Sickle cell disorders patients, especially children, have an increased risk of stroke due to the abnormal flow of blood in the cerebral arteries.
- Priapism: Priapism is a painful, prolonged erection that can occur in males with SCD. It is a medical emergency and requires immediate treatment.
- Vision Problems: Blood vessel blockages in the eyes can cause vision problems and, in severe cases, lead to blindness.
- Gallstones: Sickle cell disorders increases the risk of gallstones, which can cause abdominal pain and other digestive issues.
- Leg Ulcers: Some individuals with SCD may develop leg ulcers, which are painful sores that can be challenging to heal.
It’s important to note that the severity and frequency of symptoms can vary widely among individuals with Sickle cell disorders. Some may experience relatively mild symptoms, while others may face more frequent and severe complications. Early diagnosis and appropriate management are crucial to improving the quality of life for those living with this chronic condition.
Complications:
Sickle cell disease can lead to several serious complications, including:
- Acute Chest Syndrome: A life-threatening condition characterized by chest pain, fever, and difficulty breathing.
- Stroke: Sickle cell disorders patients are at risk of stroke, especially in childhood.
- Organ Damage: Over time, the recurrent blockages can damage organs such as the liver, heart, and spleen.
- Vision Problems: Blood vessel blockages in the eyes can cause vision problems and even blindness.
Treatment and Management:
While there is no cure for Sickle cell disorders, various treatments and management strategies can help improve the quality of life for individuals with SCD:
- Pain Management: Pain crises are treated with pain relievers, hydration, and rest.
- Hydroxyurea: This medication can reduce the frequency and severity of pain crises and other complications.
- Blood Transfusions: Periodic transfusions can increase the number of healthy red blood cells and reduce the risk of complications.
- Bone Marrow Transplantation: A bone marrow transplant from a compatible donor can potentially cure Sickle cell disorders.
- Folic Acid Supplementation: Folic acid helps in the production of red blood cells and is often prescribed to SCD patients.
- Infection Prevention: Vaccines, antibiotic prophylaxis, and regular check-ups help prevent infections.
- Hydration: Staying well-hydrated can help reduce the risk of sickle cell crises.
- Education and Counseling: Patient education and psychological support are essential for coping with the emotional and physical challenges of Sickle cell disorders.
Conclusion
Sickle cell disordersis a complex genetic blood disorder that affects millions of people worldwide. It requires ongoing medical care and management to alleviate symptoms, prevent complications, and improve the overall quality of life for individuals living with this condition. Research and advancements in treatments offer hope for better outcomes and potential cures in the future.
Read also : Exploring the Delightful Boost of the Green Tea Shot 2023